

 POLSKI
POLSKI







Introduction
Congenital chloride diarrhea (CLD) is a rare autosomal recessively inherited chronic watery diarrhea condition, which is caused by mutations in solute linked carrier family 26, member 3 (SLC26A3) leading to defective absorption of chloride in intestinal tract. However, CLD diagnosis can be confused with Batter syndrome, Gitelman syndrome and renal tubular acidosis. Neonates with CLD may appear to have watery stools (diarrhea), as well as delayed growth and development. In addition, CLD causes electrolyte imbalance, including low blood potassium levels (hypokalemia) and chlorine levels (hypochloremia). Electrolyte imbalance and severe dehydration can lead to loss of acid from the blood (metabolic alkalosis). Therefore, electrolyte replacement is a key method to control the symptoms and treat CLD. The clinical diagnosis and genetic analysis of a pediatric and their family, as well as a review of the relevant literature, are helpful to improve a clinician’s understanding of CLD.
Clinical data
The patient was a 4-month old boy admitted for anorexia, which lasted for 3 days. The patient was the second offspring and the second birth, and excessive accumulation of amniotic fluid observed during pregnancy. The patient born at 36 weeks of gestation with nature delivery, with a birth body weight of 2.8 kg, and was breastfeed. The parents were not relatives and had no history of familial disorders. On the fourth postnatal month, the patient became anorexia and had moderate diarrhea, which occurred 6–7 times per day . In addition, little mucus and blood were observed in the stool. The results of serum electrolytes from the outpatient suggested that the potassium, sodium and chloride levels were under the normal range. Therefore, the patient was admitted in the hospital.
The patients’ body weight was 4.1 kg and the blood pressure was 62/50 mmHg, while his general condition and reactions were poor. The head circumference of the patients was 40 cm and the front sputum was soft. In addition, the lips and skin of the patient were dry, the eyelids were slightly sunken and the pharynx was congested. The auscultation of the lungs identified heavy breathing sounds and there was no obvious pulmonary rales. Moreover, the auscultation of the heart had low and blunt sounds, the rhythm was clear and no heart murmur was found. The abdomen was soft and the bowel sounds were weakened, while muscle tension in the limbs of the patient was normal.
Blood gas analysis indicated metabolic alkalosis, and urine analysis revealed the presence of alkaline urine. The laboratory examination results were as follows: Potassium (K+) 2.3 mmol/l, sodium (Na+) 131.4 mmol/l and chloride (Cl–) 74.7 mmol/l. Blood gas analysis identified the pH value was 7.576, the oxygen partial pressure (PO2) was 55.6 mm Hg, partial pressure of carbon dioxide (PCO2) was 39.2 mm Hg, bicarbonate (HCO3–) was 36.5 mmol/l and the base excess (BE) was 13.0. There were four indicators of hypertension: Angiotensin I (AngI), 38.81 ng/ml; plasma renin activity (PRA), 31.48 ng/ ml/h (0.15–2.33); aldosterone (ALD), 84.43 pg/ml (10–160); and ARR, 3.27. Urinalysis results were as follows: pH value, 8.5; and the rest indexes were normal. The blood and stool routing test, adrenal gland cortex and thyroid function, liver and kidney function test, myocardial enzyme, glucose and calcifediol levels, allergens test and urine calcium/creatinine ratio were normal. The results of electrocardiogram indicated that the patient had a sinus rhythm and the right axis deviation was +211°. The results of heart doppler ultrasound identified that the foramen ovale has not been closed. Moreover, the echogenic renal parenchyma sonography, liver, gallbladder, pancreas and spleen sonography were normal.
Differential diagnosis
To identify CLD, Bartter syndrome, Gitelman syndrome and renal tubular acidosis, the history of clinical features, symptoms, signs and auxiliary examination results should be combined. Recently, there have been multiple studies regarding the aforementioned diseases; therefore, the current study summarized the etiology, pathophysiology, diagnosis and treatment of these four types of disease (Table I) [1], in order to establish clinical guidance, differential diagnosis and treatment.
Table I
Routine identification of four diseases
Bartter syndrome
The pathogenesis of Bartter syndrome is related to the dysfunction of the Na+-K+-2Cl– cotransporter in the thick ascending limb of the nephron. This disease is often onset in young children, and characterized by polydipsia, polyuria, halophilic and dehydration, as well as being accompanied by vomiting and constipation. Laboratory tests revealed a decrease in the level of serum potassium, metabolic alkalosis, increased plasma renin activity and elevated aldosterone, but without hypertension.
Gitelman syndrome
Gitelman syndrome is caused by genetic mutations resulting in the improper function of the thiazide-sensitive Na+-Cl–symporter located in the distal convoluted tubule of the kidney. The clinical manifestations include normal blood pressure, polydipsia, polyuria, limb weakness and hand and foot convulsions. Laboratory indexes of Gitelman syndrome are low levels of chloride, potassium and magnesium in the blood, as well as decreased calcium excretion in the urine. In addition, patients affected by Gitelman syndrome generally have low or normal blood pressure, high blood pH in combination with high levels of angiotensin II, renin activity and aldosterone, which could be summarized as “four lows and four highs”.
Renal tubular acidosis
Renal tubular acidosis is a medical condition that involves an accumulation of acid in the body due to a failure of the kidneys to appropriately acidify the urine. The metabolic acidosis that results from renal tubular acidosis may be caused either by failure to reabsorb sufficient bicarbonate ions (HCO3–) from the filtrate in the early portion of the nephron or by insufficient secretion of hydrogen ions (H+) into the latter portions of the nephron (the distal tubule). Renal tubular acidosis can be divided into four types; type I is manifested as polydipsia, polyuria, anorexia, vomit, diarrhea, delayed growth and development. The laboratory examination results identified chronic high chloride acidosis, hypokalemia, hyponatremia, and urine analysis demonstrated alkaline urine.
Genetic diagnosis and treatment
After admission, the patient received a positive anti-infectious treatment and electrolyte replacement to correct the electrolyte imbalance, but the therapeutic effect was poor. Low levels of potassium, sodium and chlorine, as well as metabolic alkalosis were found repeatedly. Therefore, the patient may be affected by Batter syndrome. Then, the patient received spironolactone by oral administration, indomethacin and potassium chloride sustained-release tablets. The moisture of the feces was reduced, the ingested amount of milk gradually increased and the amount of urine was moderate, thus he discharged from hospital. After being discharged for 10 days, the child was re-admitted to hospital due to “anorexia, diarrhea and fever”; the patient was re-admitted to hospitala total of three times. The patient had diarrhea > 10 times per day, accompanied with hypokalemia, hyponatremia, hypochloremia and metabolic alkalosis. To further confirm the diagnosis, genetic endocrine disease-related gene detection was performed for the patient with the family’s informed consent; Agilent exon chip capture and high-throughput sequencing technology were used.
DNA extraction
Peripheral blood samples (2 ml) were collected from the affected patient and from his parents using EDTA anticoagulant tubes. Genomic DNA was isolated from the blood sample using a rapid extraction and purification kit.
Sequencing and analysis
The capture probe of the exon of the target gene and its flanking sequences were designed, and the target fragment was obtained by hybridization and enriched by PCR amplification. The amplified product was purified and quantified, followed by high-throughput sequence using the second-generation sequencer. The sequencing of the investigated regions of the SLC26A3 revealed a homozygous mutation c.269_270dupAA (NM_000111) in exon 3. Due to the frameshift mutation, the synthetic amino acid sequence began to change from amino acid 91 (Gly). The clinically unaffected parents carried the mutations in heterozygous form, as presented in Figs. 1 and 2.
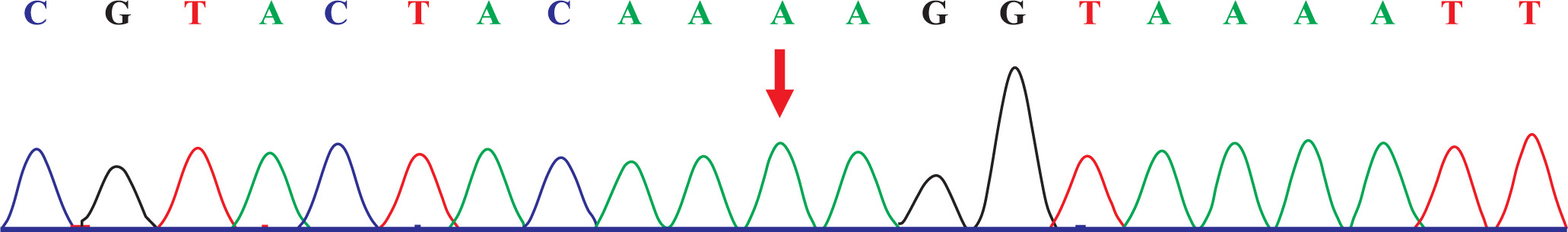
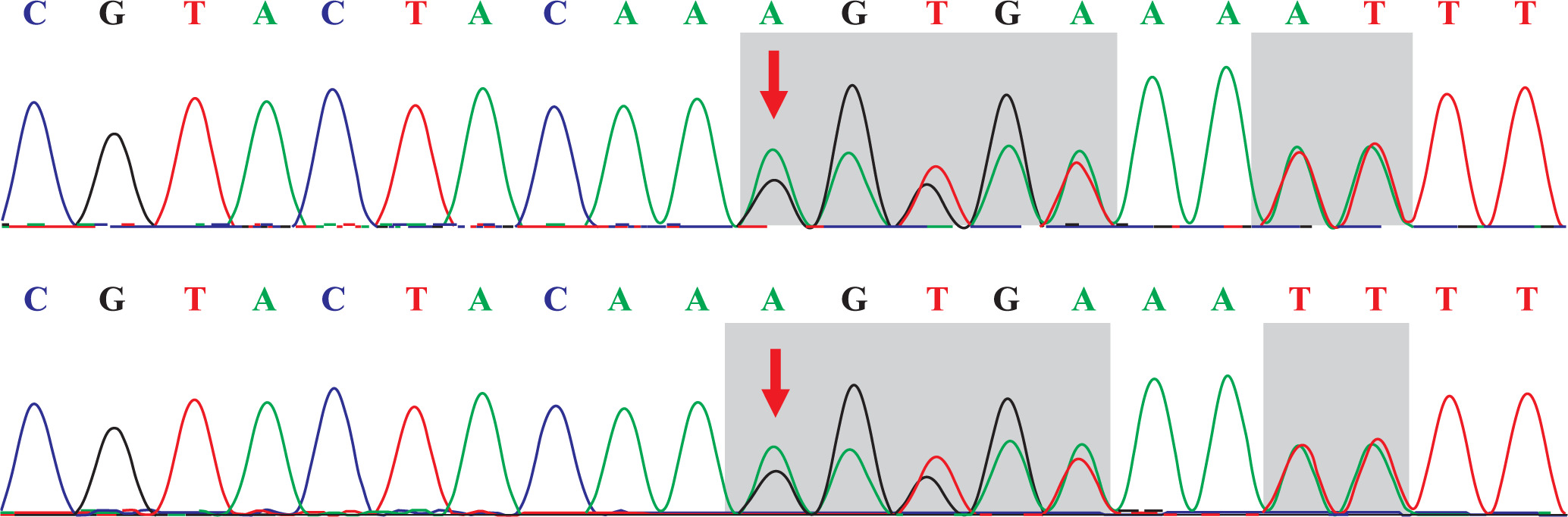
Discussion
CLD is an autosomal recessive disorder, and consanguineous mating is the primary cause. Moreover, ~250 cases have been reported worldwide, with 1/5 of the cases from Finland [1, 2]. In China, eight cases have been identified, which presented in a sporadic manner [3]. Prenatal symptoms of CLD may be manifested as prenatal polyhydramnios and bowel dilation of the fetus under ultrasound examination [4–6]. During the neonatal period, persistent watery stools, hyponatremia, hypochloremia, hypokalemia and metabolic alkalosis are the main features [7]. Furthermore, the pathogenesis of CLD is caused by the mutation of the SLC26A3 gene on chromosome 7 [8], leading to Cl–/HCO3– exchange impairment. This genetic mutation results in active transport failure of Cl– in the intestine. Elevated stool chloride is presented, and ultimately hypochloremia is observed in the affected patient. When the absorption of Cl– is inhibited, Na+/H+ exchange transport is also blocked. Therefore, the electrolyte concentration in the intestinal lumen increases, and severe osmotic diarrhea occurs. Persistent loss of fluid and sodium leads to secondary hyperaldosteronism, which could cause potassium loss. Thus, hyponatremia and hypokalemia are also observed in the affected patients [2, 7]. Collectively, it was suggested that biochemical changes are primarily hyponatremia, hypokalemia, hypokalemia and metabolic alkalosis, as well as compensatory hypercalcemia caused by metabolic alkalosis [9].
The 4-month old patient included the current study had anorexia, intractable watery diarrhea, electrolyte impairment and acid-base imbalance (hyponatremia, hypokalemia, hypokalemia and metabolic alkalosis), and was admitted to hospital three times. The patient received anti-infectious treatment, replacement of diarrhea milk powder, montmorillonite powder and probiotics by oral therapy; however, the therapeutic effect was poor. Therefore, it was speculated that the patient may not have a common infectious or non-infectious diarrhea. According to the biochemical indexes change, he was suspected to be affected by Bartter syndrome, but the symptoms of repeated diarrhea could not be explained. Finally, the diagnosis was confirmed as CLD combining the results of genetic test. Due to the low incidence of the disease and the unconventional examination of electrolytes in the feces, the disorder was easily misdiagnosed.
The clinical symptoms of Batter syndrome and CLD are confounded. With regards to Batter syndrome [10], the dysfunction of the Na+-K+-2Cl– cotransporter in the thick ascending limb of the nephron of kidney is the main feature, which results in chloride loss from urine. In contrast, the pathogenesis of CLD includes the impaired active transport of Cl–/HCO3– in the ileum and colon, which leads to chloride loss from the stool. Therefore, to identify Batter syndrome and CLD, it is necessary to detect the urine and feces electrolyte levels. A main feature of CLD is that the chloride level is higher compared with the sum of the sodium and potassium levels; the stool chloride level is > 90 mmol/l after the treatment [11–14]. Without therapy, the stool chloride level is < 40 mmol/l, suggesting severe dehydration [15]. Therefore, it is helpful for diagnosis to detect the electrolyte level in the stool repeatedly after correcting dehydration. Genetic testing is the most effective method for the diagnosis of CLD. Currently, > 50 mutations of the SLC26A3 gene have been reported [16]. In the present study, the affected patient had a homozygous mutation of c.269_270dupAA (NM_000111), resulting in the frameshift mutation of the amino acid. The parents of the patient carried the mutations in the heterozygous form at the same position. In addition, this mutation has been revealed to be pathogenic [16].
Maintaining the electrolyte and acid-base balance is a major therapeutic method for CLD, but it is difficult to treat diarrhea [11, 16]. Replenishment of KCl and NaCl is the most effective method for the treatment of CLD, and combined treatment demonstrates an improved therapeutic effect compared with the single application of KCl. In the early neonatal stage, the administration route should gradually move from intravenous to oral rehydration fluid. Moreover, the recommended replacement doses of chloride are: Young child, 3–5 mmol/ (kg•d; NaCl : KCl, 6 : 5); infant, 6 mmol/ (kg • d; NaCl : KCl, 2 : l); and newborn, 6–8 mmol/(kg • d; NaCl : KCl, 3 : 1). Maintaining the urine chloride level in the normal range, as well as the electrolyte and acid-base balance are the hallmarks of adequate treatment. However, due to the mutation of SLC26A3 in the intestinal mucosa, diarrhea persists. Omeprazole, cholestyramine and butyrate are prescribed to the affected pediatric patients, but the therapeutic effect on reducing the frequency of diarrhea shows individual variety. Failure to diagnose and treat the disease early can lead to repeated intestinal infection, fluid and electrolyte disorder, and eventually causing delayed growth and development. The content of NH4+ increases along with age, and can combine with hydrogen ions to promote the absorption of sodium; therefore, the dose of NaCl can be reduced gradually, while potassium and chloride continue to be excreted, and thus it is necessary for the patient to receive KCl replacement [9, 16]. However, due to the low incidence of CLD, large-scale clinical trials cannot be performed, which hinders the development of novel effective therapeutic methods.
In conclusion, CLD can seriously affect the growth and development of pediatric patients, and failure to diagnose and treat in a timely manner can be life-threatening. CLD diagnosis should be considered with the detection of excessive accumulation of amniotic fluid during pregnancy and newborn who present with watery diarrhea, hyponatremia, hypokalemia, hypokalemia and metabolic alkalosis. In addition, genetic tests should be conducted to achieve early diagnosis and treatment. Timely diagnosis and improving the prognosis effective could also be achieved by comparing findings from reported incidences worldwide.