

 POLSKI
POLSKI







Introduction
Cushing’s syndrome (CS) is a rare disease in childhood. Paediatric CS differs from that in adults in various aspects. The classical features of protein catabolism seen in adult CS are usually absent in children. In adults, pituitary adenoma is the most common cause of endogenous CS, while in the paediatric age group, adrenal causes predominate over other aetiologies. Adrenocortical carcinoma remains the most common aetiology between 1 and 5 years of age [1]. Most adrenocortical carcinomas co-secrete other hormones, such as testosterone, 17-hydroxy-progesterone [17(OH)P], deoxy-corticosterone acetate (DOCA), or even aldosterone in addition to cortisol [2]. This usually manifests as gonadotrophin-independent precocious puberty in addition to CS. Concomitant precocious puberty may often mask the symptoms of cortisol excess. Benign adrenal adenomas constitute a rare aetiology for paediatric CS. It is rare for benign tumours to co-secrete other hormones apart from cortisol. Here, we describe a case of Cushing’s syndrome with precocious puberty in a toddler due to benign adrenocortical adenoma co-secreting cortisol and testosterone.
Case report
A 2.5-year-old male child, born of non-consanguineous marriage, had normal antenatal, intranatal, and postnatal histories. He presented to the endocrine clinic with the complaint of 11 kg weight gain over a duration of 6 months. This was associated with increased appetite. There was marked prominence of the dorso-cervical pad of fat. There was also an increase in the circumference of his arms and legs. There was no recent acceleration or deceleration of height. His parents also noticed a recent change in the behaviour of the child with increased aggressiveness and hoarseness of voice. There was increase in the size of the phallus and nocturnal penile tumescence. There was also the appearance of axillary and pubic hair. However, there was no history of acneiform eruptions or malodorous secretions.
He had no history of bruising, ecchymosis, striae, or proximal myopathy. There was no history of exposure to any exogenous medications. There was no history of headache, visual impairment, or seizures. There was no history of any bone deformity or swelling or any pigmented skin lesion to suggest McCune-Albright syndrome, which can be a cause of CS in infancy. There was also no history suggestive of any syndromic cause of CS like spotty skin pigmentation, skin lentigines, any nevus or testicular tumour (Carney complex); any neuroglycopenic symptom to suggest hypoglycaemia as in insulinoma; any graveluria, flank pain to suggest hypercalciuria as in primary hyperparathyroidism (Multiple Endocrine Neoplasia type 1).
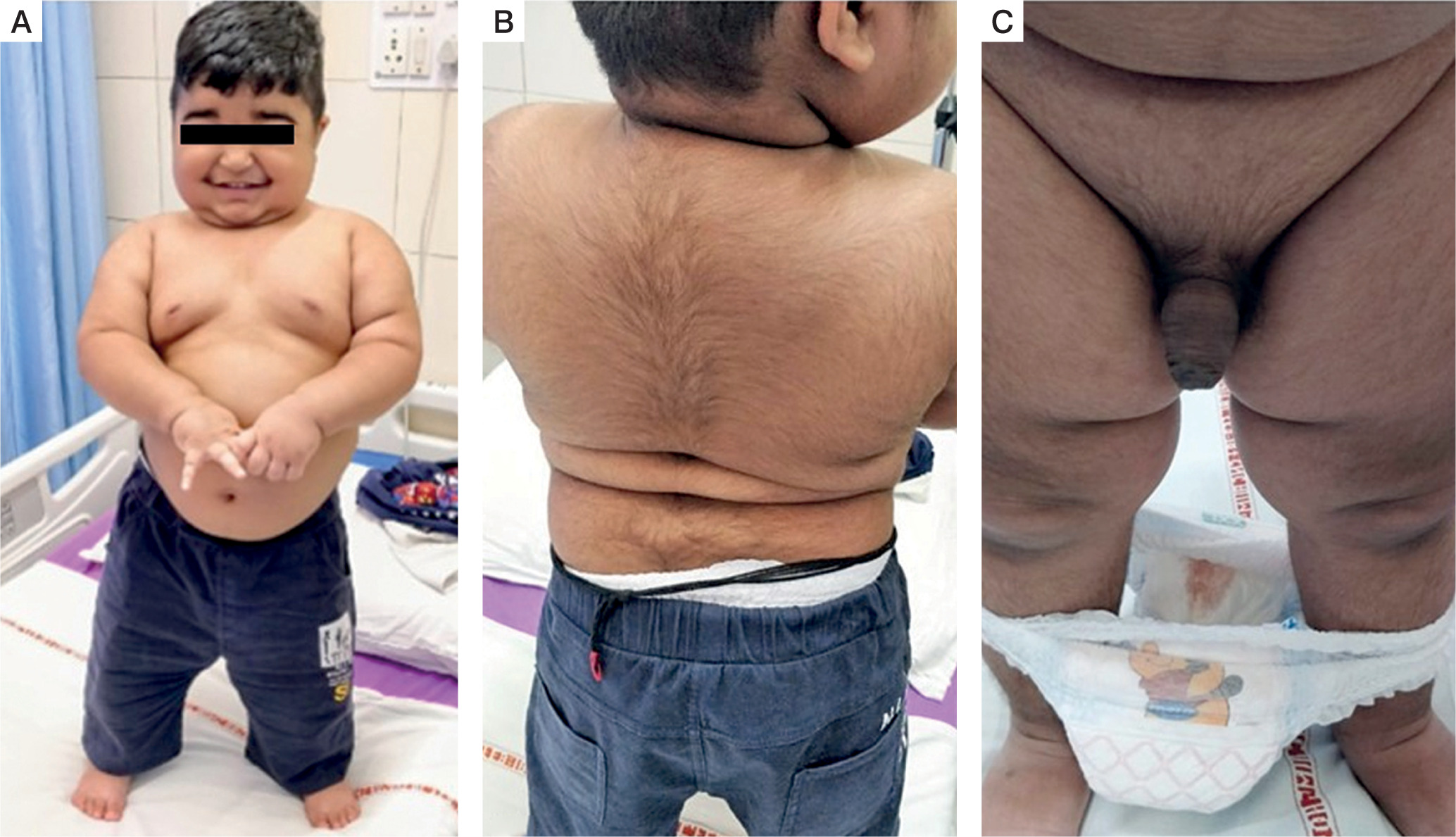
A physical examination revealed a height of 92 cm, which corresponded to the 50th centile, and weight of 22 kgs, which corresponded to more than the 97th centile, as per Indian Academy of Paediatrics reference data for Indian children [3]. The patient was hypertensive with both systolic and diastolic blood pressures more than the 90th centile, as per the Indian Academy of Paediatrics percentile charts for blood pressure [4]. He had rounding of the face, facial and truncal hypertrichosis, acanthosis nigricans grade II, prominent dorso-cervical pad of fat, along with central obesity (Fig. 1A, B). He had a waddling gait. Testicular volume was 2 ml bilaterally, with stretched penile length (SPL) of 7 cm. SPL was more than the 95th centile for the age of 2.5 years [5], as per the reference data for Indian children. Pubic hair stage was P3 (Fig. 1C).
Figure 1
Clinical features of a 2.5 year old male patient with pediatric Cushing’s syndrome, and pseudo-precocious puberty: A, B) roundening of face, truncal hypertrichosis, acanthosis nigricans grade II, prominent dorsocervical pad of fat along with central obesity; C) increased phallic length, and pubic hair stage of P3
Investigations revealed normal haemogram, and liver and kidney function tests, with no evidence of hypokalaemia. Hormone profile suggested ACTH-independent Cushing’s syndrome (Table I). His morning serum cortisol was 24.2 µg/dl (normal, 6.9–19.4 µg/dl), 11 p.m. serum cortisol was 22.4 µg/dl (normal < 7.5 µg/dl), and 11 p.m. salivary cortisol was 1.4 µg/dl (normal < 0.4 µg/dl). His low-dose dexamethasone suppression test revealed a non-suppressed cortisol level of 24.1 µg/dl (normal < 1.8 µg/dl). His plasma adrenocorticotrophic hormone (ACTH) was less than 1 pg/ml (normal 5–15 pg/ml). Hormone profile also revealed gonadotrophin release hormone (GnRH)-independent precocious puberty, with suppressed luteinizing hormone (LH) and follicle stimulating hormone (FSH) (LH < 0.1 mIU/ml, FSH < 0.1 mIU/ml). Total testosterone was elevated for his age, at 1.93 ng/ml (normal < 0.2 ng/ml). Among other adrenal hormones, serum dehydroepiandrosterone sulphate (DHEAS – 232.4 µg/dl, normal < 40 µg/dl) and serum 17 hydroxy progesterone (17[OH]P – 1.6 ng/ml, normal range, 0.16–0.74 ng/ml) were raised. Thyroid function tests were within normal limits. The bone age of the child measured by Greulich and Pyle atlas was 2.5 years, corresponding with the chronological age [6].
Table I
Hormonal parameters of our patient before and after 6 months of surgery
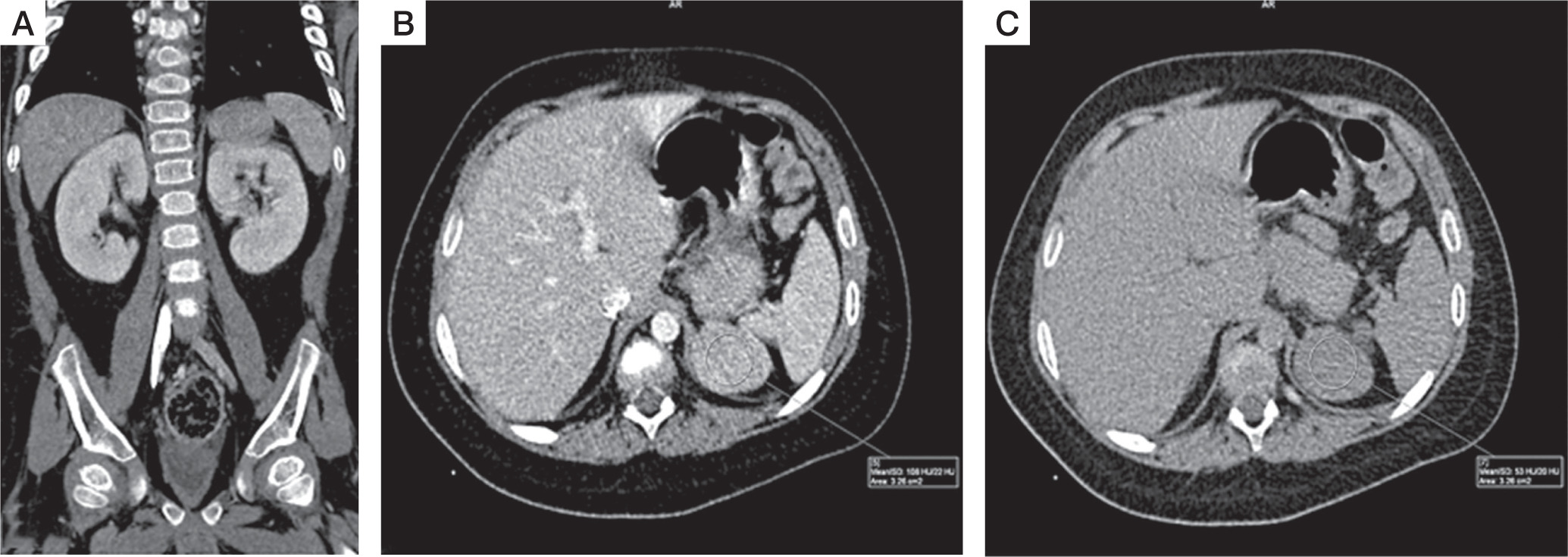
He was then subjected to computed tomography (CT) of the abdomen, which revealed a large 3.9 cm left suprarenal mass with smooth, well defined margins suggestive of adrenocortical adenoma. (Fig. 2A–C). Non-contrast CT was not performed, so non-contrast attenuation was not available. Post-contrast CT showed an attenuation of 108 Hounsfield units (HU) in the portal venous phase and 53 HU on 15-minute delayed phase. Post contrast, relative washout was 51%, again suggesting adrenocortical adenoma. There was no calcification, necrosis, or metastasis evident on imaging.
Figure 2
CT features suggestive of adrenal adenoma: A) coronal and figure; B) axial portal venous phase contrast enhanced CT image; C) axial 15 minuts delayed contrast enhanced CT image shows well-defined left adrenal mass, with attenuation of 108 HU on portal venous phase, and 53 HU on 15 minuts delayed phase, and relative washout of 51%
He was operated under the cover of perioperative intravenous glucocorticoids, and the tumour was resected with the capsule intact. Histopathology revealed a moderately circumscribed tumour measuring 3 ´ 1.5 ´ 1 cm. The tumour grossly weighed 15 g. No necrosis or calcification was identified. Multiple sections showed an encapsulated tumour, composed of polygonal cells with abundant eosinophilic cytoplasm, round nuclei, and large prominent nucleoli (Fig. 3A–D). The cells were arranged as trabeculae and sheets and showed mitotic activity of 5–6/20 high-power fields. Capsular or vascular invasion or atypical mitoses was not seen. The tumour cells were focally positive for inhibin and negative for chromogranin and Melan-A (Fig. 4A, B). Normal adrenal gland was seen at the periphery. Using Wieneke criteria [7], features were compatible with adrenocortical adenoma (Table II). Ki-67 index was low, i.e. 2% in the areas of the highest mitotic activity.
Figure 3
A–D) Histopathology of left adrenal mass (hematoxylin and eosin stain), showing encapsulated tumor, composed of polygonal cells with abundant eosinophilic cytoplasm, round nuclei and large prominent nucleoli that are arranged as trabeculae and sheets and showed mitotic activity of 5–6/20 high power field. No capsular or vascular invasion or atypical mitoses was seen
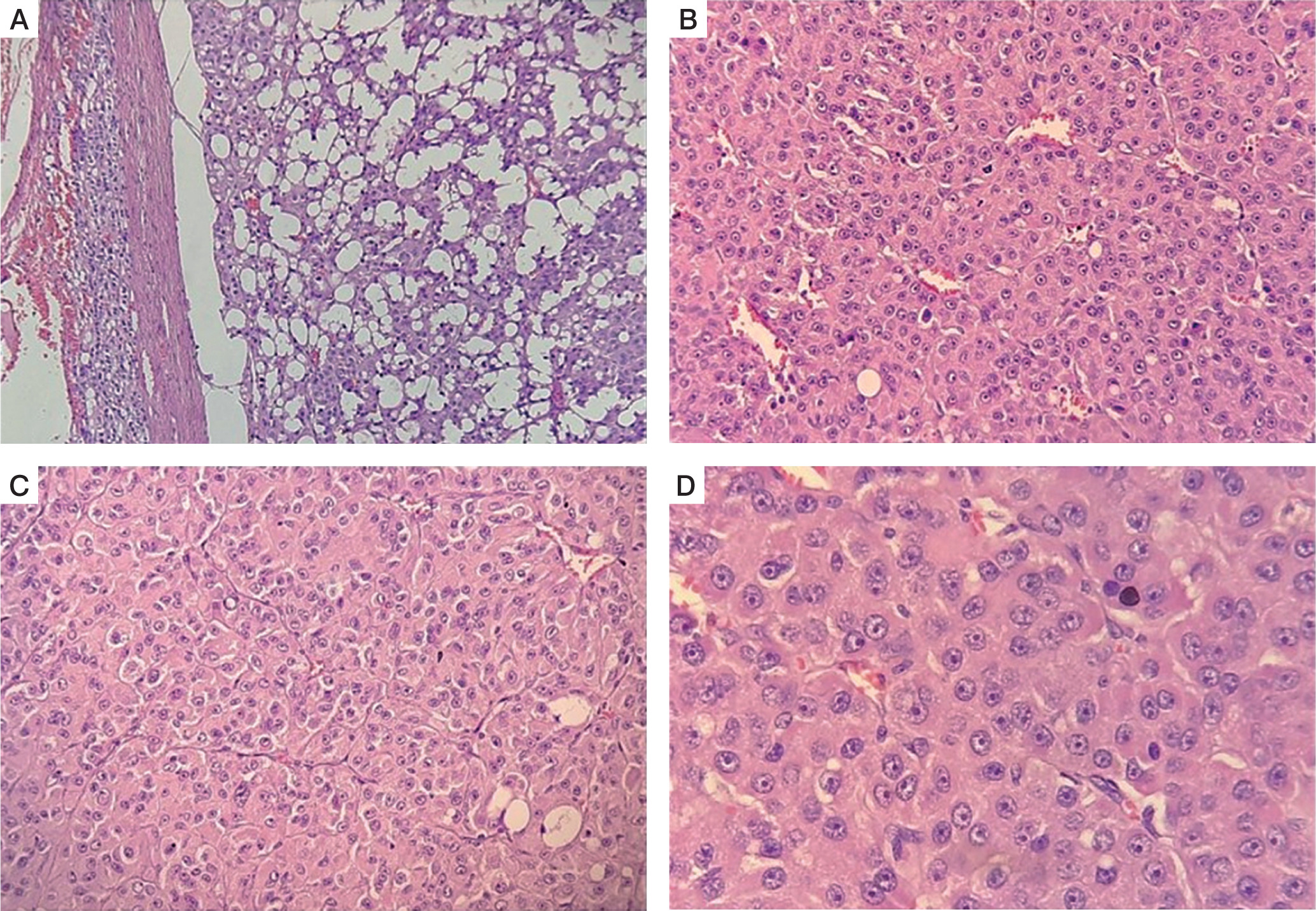
Figure 4
A, B) Immunohistochemistry shows that tumor cells were focally positive for inhibin and negative for chromogranin and melan A
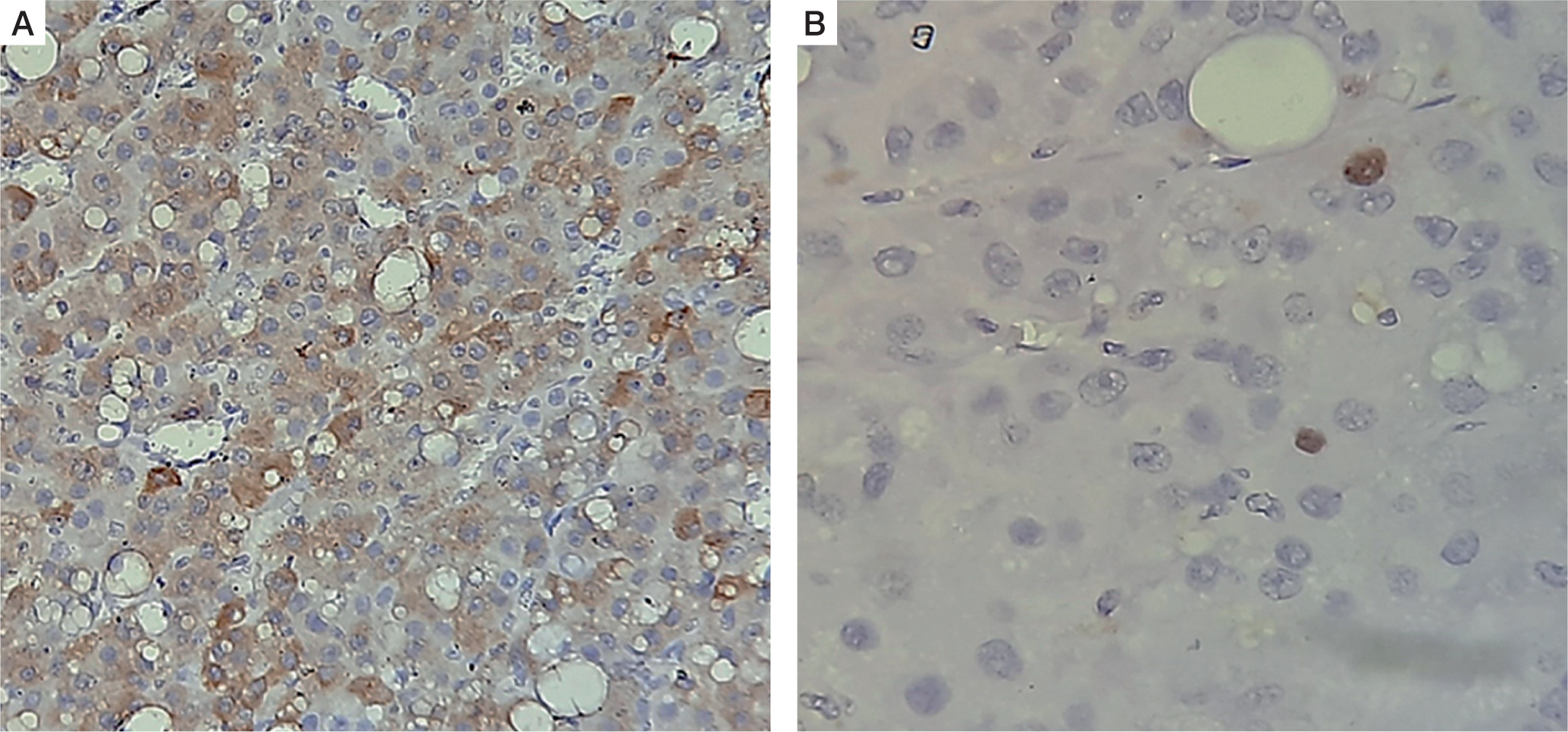
Table II
A comparative analysis of Wieneke scoring criteria and the histopathological findings in our case
After surgery, the patient was switched to oral glucocorticoids. Vitals and blood glucose were monitored in the post-operative period. He was discharged on tablet hydrocortisone 5 mg at 8 a.m., 2.5 mg at 1 p.m., and 2.5 mg at 6 p.m. At the time of writing, the patient had completed 6 months of successful surgery. He lost 10–11 kgs of weight. Biochemical assessment after 6 months revealed serum cortisol < 0.054 µg/dl (after omitting glucocorticoid for 24 hours), serum LH < 0.1 mIU/l, and serum testosterone < 0.025 ng/ml (Table I). Serum DHEAS had also fallen to 0.215 µg/dl. There was no increase in testicular or phallus size or increase in the distribution or density of axillary or pubic hair.
Discussion
Cushing’s syndrome is a rare disease in childhood. In our patient, the clinical and biochemical manifestations were suggestive of cortisol excess and co-secretion of androgens. Involvement of more than one steroidogenic pathway, as in our case, leads to the suspicion of adrenocortical carcinoma [2]. Most carcinomas have inefficient steroid synthesis and co-secrete steroid precursors like deoxycorticosterone acetate (DOCA), 17-hydroxyprogesterone [17(OH)P], dehydroepiandrosterone sulphate (DHEAS), aldosterone, and androgens such as testosterone [8]. In our patient, 17(OH)P, DHEAS, and testosterone were elevated, but DOCA and aldosterone levels were not measured.
On imaging, the CT characteristics were not consistent with adrenocortical carcinoma (ACC). The suprarenal mass had homogenous enhancement, regular margins, and relative washout of 51% with no necrosis or calcification, suggesting adrenocortical adenoma. Non-contrast attenuation was not available with us, while post-contrast attenuation was quite high (108 HU) in the portal venous phase. Such high portal venous attenuation can be encountered in pheochromocytomas [9], but the clinical and biochemical picture suggested against it. Several studies have now shown that the absolute attenuation value in the post-contrast venous phase does not allow reliable differentiation between benign adrenocortical adenomas and other lesions like pheochromocytomas and adrenocortical carcinomas [10, 11]. Szolar et al. reported that adenomas exhibited a much higher (almost 10 times) mean enhancement from the baseline after 60 seconds of contrast administration, a finding due to lower pre-contrast attenuation of adenomas [10]. The authors concluded that given the overlapping venous phase enhancement patterns of adenomas and non-adenomas, it could not reliably differentiate adenomas from other lesions. Therefore, given the bigger size of approximately 4 cm, ACC still could not be conclusively ruled out.
Peri-operatively, the patient was started on glucocorticoids that were continued in the post-operative period. It is important to administer glucocorticoids because the contralateral adrenal gland may remain suppressed for a long time, sometimes even requiring more than a year for recovery [12]. The suppression takes place at the level of the hypothalamus (hypothalamic pituitary adrenal [HPA] axis) due to excess cortisol produced by the tumour. Recovery of the HPA axis should be assessed every 5-6 months. After 24 hours of the last dose of hydrocortisone, an ACTH stimulation test is to be done by injecting intravenous ACTH 250 mg (Synacthen 250 mg IV). If the peak cortisol level after 1 hour of Synacthen injection is more than 18 µg/dl, then glucocorticoids can be safely withdrawn [13].
Histopathology revealed that the tumour cells were surrounded by an intact capsule with normal adrenal gland seen at the periphery. Even histopathological differentiation between adrenocortical adenomas and carcinomas may be difficult [14]. To address this issue, various histopathological diagnostic scores have been used. The Weiss score is commonly used in adults [15, 16]. Unfortunately, histopathological classification in paediatric adrenocortical tumours has been unreliable, and the Weiss score has not been found to be a useful tool in children [17]. In paediatric adrenocortical tumours, the Wieneke scoring system [18] has been proposed to predict malignancy. This has been validated in a few studies [19–21]. On application of Wieneke criteria in our case, the features were consistent with adrenocortical adenoma. A comparative analysis of the Wieneke scoring system and the histopathological characteristics of our case is mentioned in Table II. Unfortunately, the tumour specimen could not be stained for TP53 mutation, which is commonly associated with adrenocortical carcinoma, as part of Li Fraumeni syndrome (a dominantly inherited familial cancer syndrome) [22]. However, a plan was made to screen the patient at regular intervals to observe for any symptoms or signs of recurrence. We also plan to perform whole-exome sequencing in the child to look for TP53 mutation and IGF2 overexpression to predict the risk of malignancy.
Post surgery, there was drastic improvement in symptoms, with 10 kg weight loss in a period of 6 months. There was also restoration of blood pressure to normal centiles. After removal of the tumour, there is sudden withdrawal of the sex steroids. This may lead to reactivation of the hypothalamic pituitary gonadal axis, causing central precocious puberty (CPP) [8]. The latter may be suspected if there is a sudden acceleration of height or increase in testicular volume. Biochemically, CPP may be confirmed on an elevated basal LH level (> 0.3 mIU/l) and stimulated LH > 5.0 mIU/l on GnRH stimulation test [23]. In our patient, there was no clinical or biochemical parameter to suggest development of CPP after 6 months of successful surgery. However, this needs regular surveillance, and GnRH analogues may be started if there is any sign of CPP.
Conclusions
Our case is a classical representation of the diagnostic uncertainty that exists in cases of adrenocortical tumours. The patient’s clinical and biochemical presentation pointed to adrenocortical carcinoma because there was involvement of more than one steroidogenic pathway. Radiological findings were suggestive of adrenocortical adenoma, but carcinoma could not be conclusively ruled out given the large size of the mass. The patient underwent surgery, and the task of discerning malignancy now relied on histopathology findings. Even histopathology may pose a difficulty in differentiating adrenocortical adenomas from carcinomas, particularly in the paediatric age group. In this regard, certain diagnostic criteria may be helpful. In our case, the Wieneke score was applied, which clearly suggested adenoma. There was resolution of symptoms after surgery. However, a plan was devised to closely follow the patient to look for any sign of relapse. This case conveys the message that there can be disparity among clinical, radiological, and histopathological aspects of adrenal tumours. It is, therefore, prudent to not completely depend upon only one entity to ascertain the character of an adrenal tumour.